作者:神經(jīng)內(nèi)科 黃剛
奔走跑跳����,是人類天生的本能��。然而,有些人走路晚���、走路慢�����,容易摔倒��。他們一生中都要和這種罕見病做斗爭��。近日����,漯河市中心醫(yī)院收治了一例主因“雙下肢行走發(fā)軟����、易跌倒20年,加重3年”的32歲男性����。經(jīng)過神經(jīng)內(nèi)科、肌電圖室��、病理科�、磁共振科等多學(xué)科會診����,完善心肌酶學(xué)��、肌電圖����、肌肉活檢和病理、肌肉磁共振����、基因檢測及家系分析等檢查后,該患者最終被確診為Becker型肌營養(yǎng)不良�。在進(jìn)行多器官系統(tǒng)全面綜合評估后,制定合理治療方案和隨訪計(jì)劃�����,患者及家屬滿意出院����。
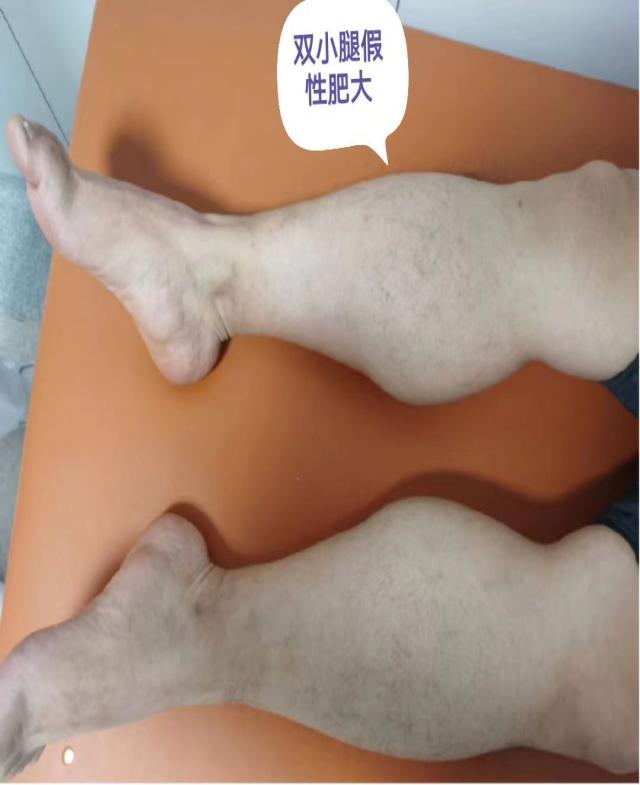

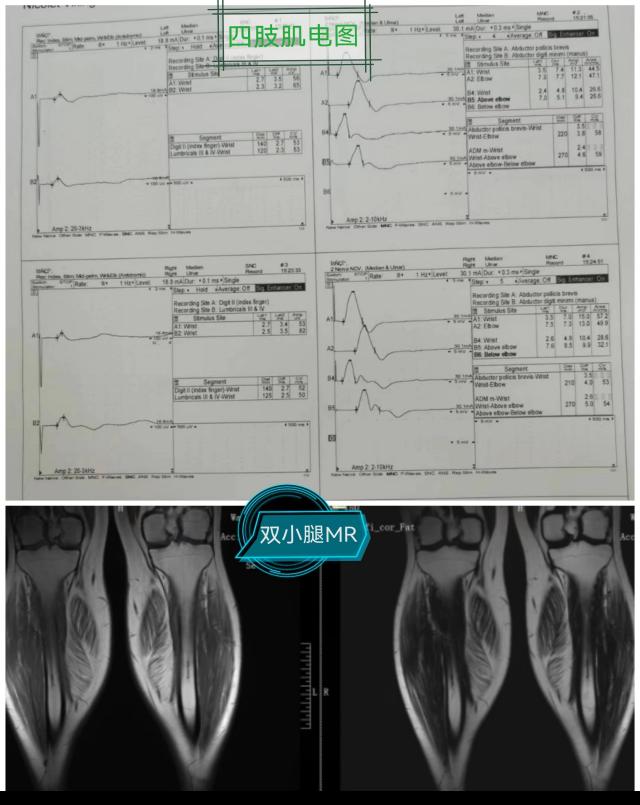
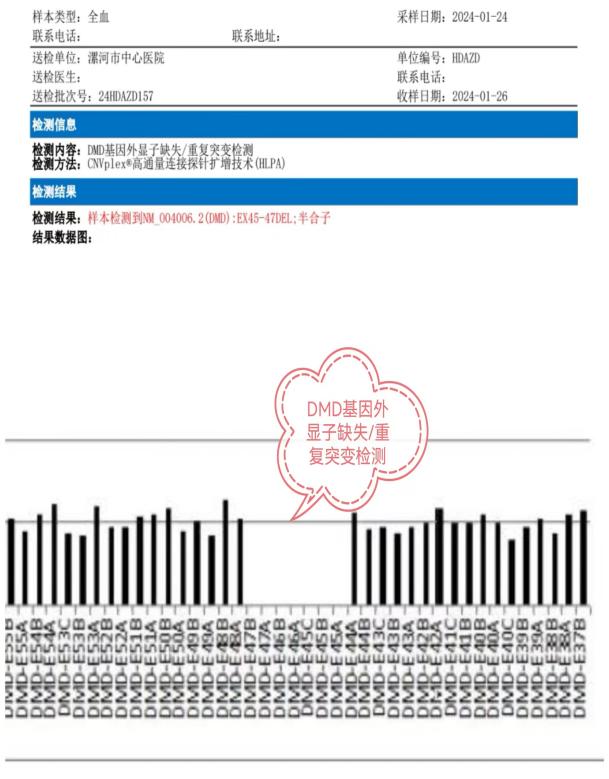
該例罕見病的確診在彰顯中心醫(yī)院診治神經(jīng)疑難病癥多學(xué)科協(xié)助綜合實(shí)力的同時(shí),也有力地促進(jìn)了神經(jīng)內(nèi)科(周圍神經(jīng)?����。﹣唽?频慕ㄔO(shè)�����,并提高了綜合醫(yī)院的教學(xué)水平���。接下來,就一起認(rèn)識一下��,什么是假肥大型肌營養(yǎng)不良吧�!
1 概念
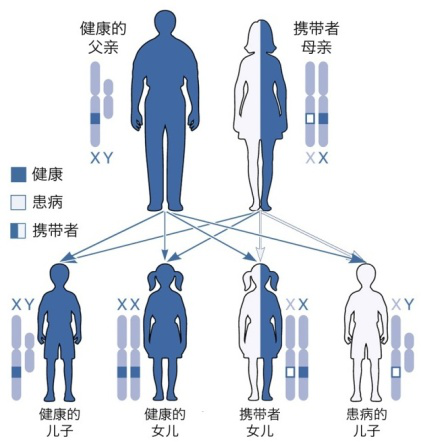
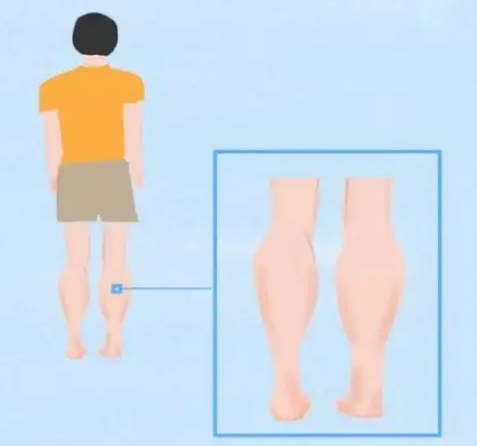
假肥大性肌營養(yǎng)不良,包括Duchene型肌營養(yǎng)不良癥(DMD)和Becker型肌營養(yǎng)不良癥(BMD),是全球最常見也是最嚴(yán)重的兒童遺傳病之一�。DMD呈X連鎖隱性遺傳,因此����,絕大多數(shù)患者都是男性。主要臨床表現(xiàn)為進(jìn)行性加重的四肢近端肌��、腰帶肌無力���、萎縮�����,腓腸肌肥大����,嚴(yán)重影響患者的日常運(yùn)動能力,病程晚期可累及呼吸肌和心肌���。
它發(fā)病率為1/3500�,我國每年約有400-500例DMD患兒出生�,累計(jì)約7萬人確診為DMD。
‎
2 發(fā)病機(jī)制
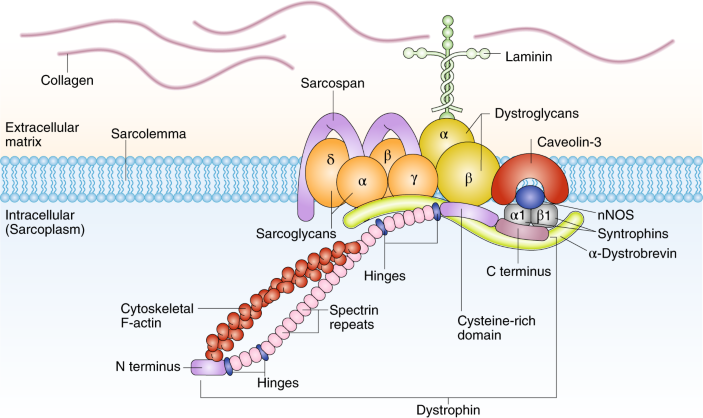
DMD的致病基因?yàn)榭辜∥s蛋白基因(dystrophin)�,位于染色體Xp21.2區(qū),全長約2.2Mb�,共包含79個(gè)外顯子,是已知最大的人類基因��?���?辜∥s蛋白主要位于骨骼肌和心肌細(xì)胞膜的質(zhì)膜面,具有細(xì)胞支架、抗?fàn)坷?����、防止肌?xì)胞膜在收縮活動時(shí)撕裂的功能。
DMD 患者因基因缺陷而使肌細(xì)胞內(nèi)缺乏抗肌萎縮蛋白,造成肌細(xì)胞膜不穩(wěn)定并導(dǎo)致肌細(xì)胞壞死和功能缺失而發(fā)病�,肌纖維的完整性遭到破壞,嚴(yán)重者可見纖維結(jié)締組織和脂肪組織替代正常的肌肉組織���,出現(xiàn)肌肉假性肥大的典型表現(xiàn)��。
3 臨床特征
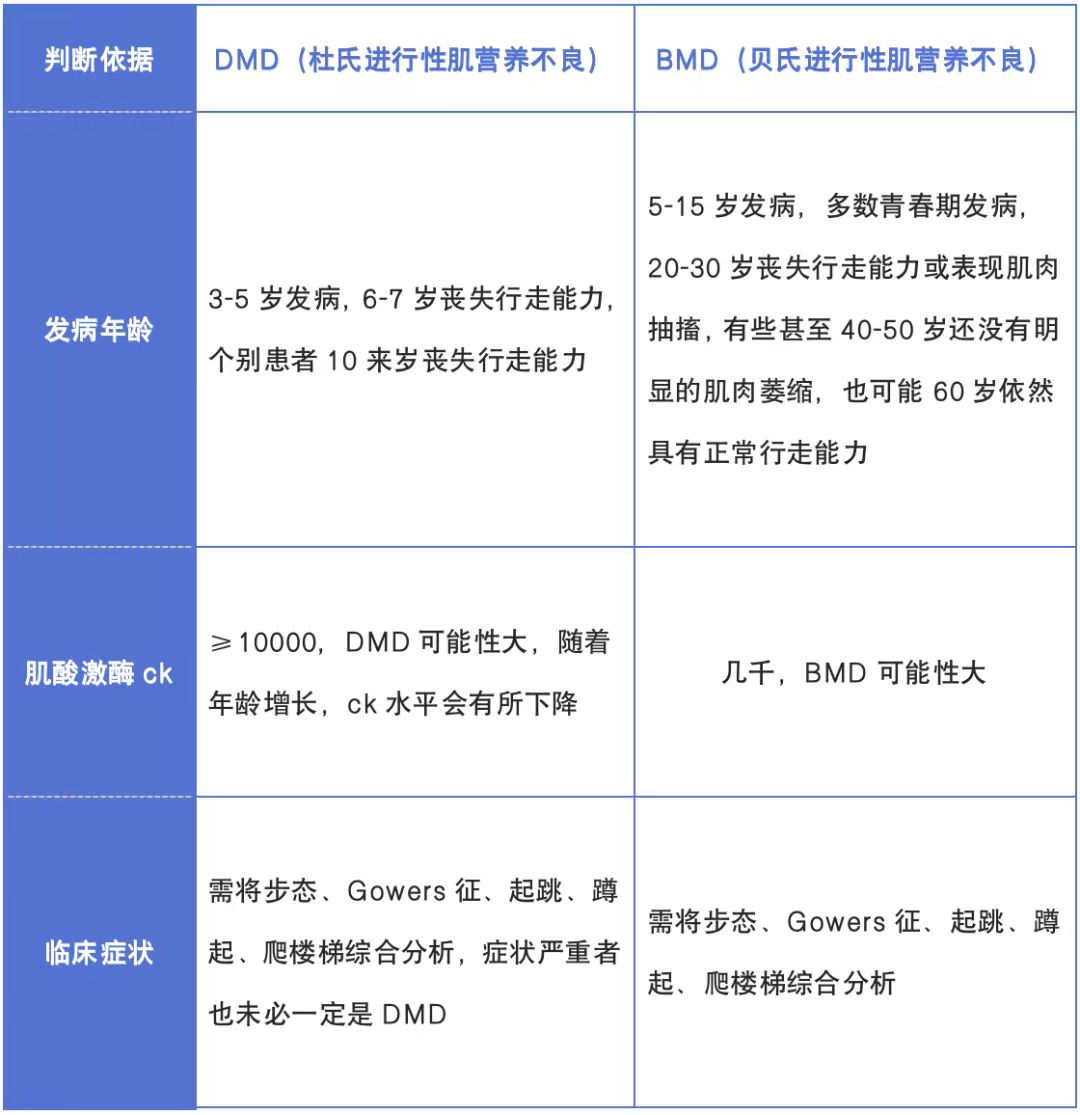
(1)Duchenne型肌營養(yǎng)不良
幼兒期運(yùn)動發(fā)育輕度遲滯�����,兒童期(5~6歲)運(yùn)動能力開始下降��,并出現(xiàn)步態(tài)異常���、跟腱攣縮��、腰椎前凸等變化����。骨骼肌進(jìn)行性無力萎縮��,影響肢體運(yùn)動功能�,逐漸出現(xiàn)步態(tài)異常、上肢活動受限,自然病程常在10歲左右喪失行走能力�。長期隨訪顯示,DMD患者大多數(shù)活不過20歲�����,就會因心肺功能衰竭而死�。
(2)Becker型肌營養(yǎng)不良
為同一疾病的相對良性表型���,發(fā)病率為DMD的1/10���。因DMD基因功能未完全喪失,所以病情明顯輕于Duchenne型肌營養(yǎng)不良����。可青年甚至成年起病�,假肥大體征明顯,基本不影響生存期����,智力正常。
4 輔助檢查和診治流程
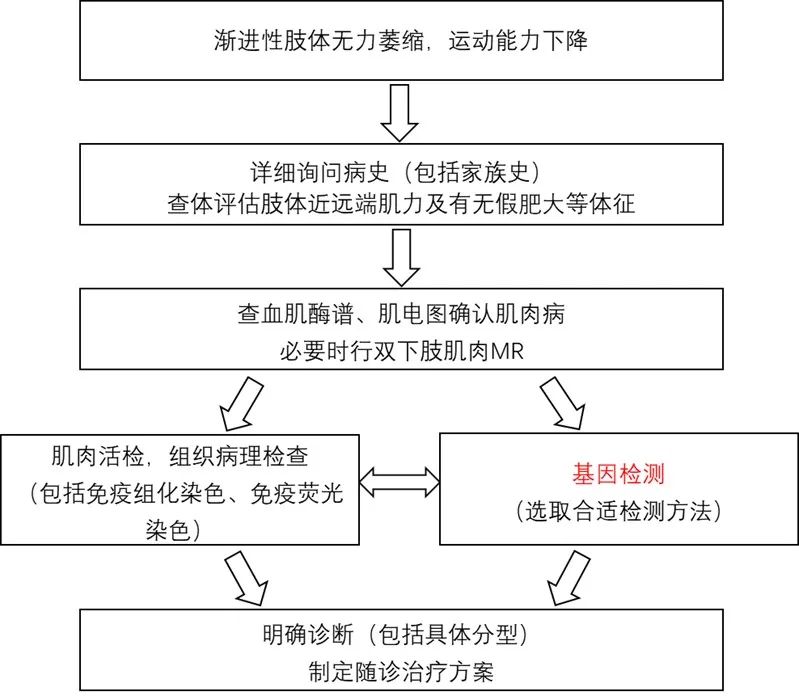
DMD和BMD的診斷相關(guān)輔助檢查包括:血清學(xué)檢測����、肌電圖����、肌肉MRI�、肌肉活檢和基因檢測,確診需基因檢測發(fā)現(xiàn)DMD基因致病性缺陷或肌肉活檢發(fā)現(xiàn)Dystrophin蛋白異常�����。

5 治療
迄今為止��,DMD尚無有效的根治方法����,但是及時(shí)的對癥支持治療和恰當(dāng)?shù)淖o(hù)理可以提高患者的生活質(zhì)量和延長壽命����。應(yīng)鼓勵患者盡可能從事日常活動,避免長期臥床��。藥物可選用小劑量激素��、ATP�����、肌苷、維生素E等���。
DMD的治療策略的研發(fā)是與疾病賽跑的過程���,對于肌營養(yǎng)不良這種疾病來說可以采用基因療法進(jìn)行治療,而致病基因鑒定是實(shí)現(xiàn)治療的第一步�,通過基因檢測明確致病基因,針對性的防治��,幫助后代不再患?����?����!基因治療(外顯子跳躍�����、微小基因替代)及干細(xì)胞移植治療改變了這些曾經(jīng)無法治愈的遺傳疾病的治療前景����,為有意義的治療干預(yù)帶來新的希望����!











